Plate-based SLAP
We recently published the paper High-throughput synthesis provides data for predicting molecular properties and reaction success. I will use this blog post to clarify a few ideas from the paper in simple terms.
Overview
So what is the advance, the novelty in this paper?
We demonstrate a high-throughput synthesis platform for saturated N-heterocycles. This includes:
- Easier/faster access to privileged structures
- Automated purification and analysis of the products to obtain lots of physicochemical property data.
- A virtual library of products accessible from commercial building blocks
- Property predictions for the virtual library
- Reaction outcome predictions for the virtual library (i.e. accessibility of the virtual library members)
Parallelization

Notice the fans for air cooling
on the bottom and the water-cooled
aluminum heat sink on the top
The first thing we did is, we took a reaction that was previously discovered by our group called SLAP (for SiLicon Amine Protocol), and we made it run in parallel. What can be summed up in this simple sentence is actually a formidable optimization challenge. The reaction comprises several steps, including purification of intermediates and a photochemical cyclization.
The details are in the paper, but to just break down one of the challenges: How do you evenly and intensely irradiate 96 vials at the same time? Moritz Jackl, who is a co-author on the paper, solved this by constructing an array of 96 blue LEDs, with 1030 mW each. Now if you do the math, it adds up to about 100 W for the entire array. That is a whole lot of heat to dissipate. So the design we finally came up with uses not just active air cooling on the bottom of the array, but also water cooling on the top, through an aluminum heat sink that the vials are fitted into.
I would love to say no LEDs were harmed in constructing this photoreactor, but you can imagine what happened until we figured out the cooling. As a result however, we get intense blue light, evenly over the whole plate, and we show that the reaction proceeds equally, independent of the position of the vial on the array.
What about all these properties?
While I could write for a long time just about the synthesis, our work didn’t stop here. Our in-house analysis capabilities for plates are largely limited to LC-MS (which is already very useful), so we partnered with the Microcycle team at Novartis who have built an automated purification system.
Through their platform, they analyzed almost a thousand reaction mixtures and, using high-throughput prep-HPLC, isolated almost 500 distinct compounds (including some diastereomers). And they didn’t stop there either. For all compounds that we obtained enough pure material of (a few mg are enough), they conducted physicochemical assays to determine log D, pKa and aqueous solubility.
Why is this important? Just think about possible applications of this platform. The molecules that can be obtained through SLAP chemistry have desirable properties from a medicinal chemistry perspective. Saturated N-heterocycles are already frequently used to improve solubility of compounds, however the available substitution patterns (mostly N-substitution) limit tunability. With our method, substitutions adjacent to the nitrogen atom are possible. In addition, being able to make many derivatives quickly, allows to tune the properties to the desired point.
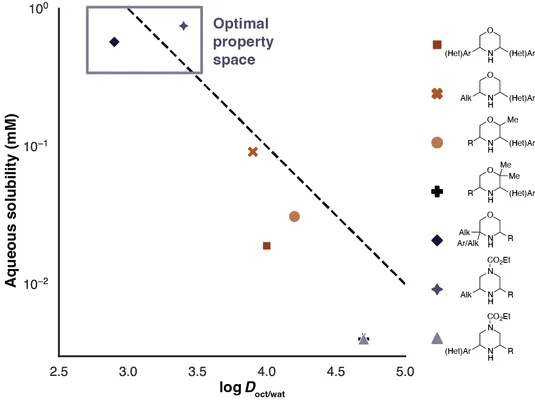
This endeavor will be expensive if we are just blindly trying anything the comes to mind. So in order to choose more rationally what to try, it is useful to have an idea about the structure property relationships for these compounds. While our measured properties allow that, we went a step further and predicted the properties for many conceivable SLAP products using machine learning.
Are the ML predictions meaningful?
Not only did we predict properties for the virtual library (VL) of possible SLAP products, we also predicted reaction outcome, i.e. whether the VL members are accessible using our platform. There has been some work on reaction outcome prediction in the past, but we found the common evaluation methods lacking. In machine learning, it is easy to end up with a model that looks good on paper, i.e. gives you nice metrics, but does not “learn” anything about chemistry. In one of the more extreme examples of this fallacy, dependencies in the data were ignored and the resulting model used the dependency structure of the data to make its predictions, not the chemical information.
This is not yet a problem. The problem only occurs once you start to ask for predictions for things the model has not seen. So the key to properly evaluate models is to be aware of what it will be used for in the future.
A closer look at data splits
Let’s take this to our SLAP platform. We have a reaction with basically two components (a SLAP reagent and an aldehyde). Of course, the SLAP reagent is produced from other things in our platform, but these steps are fully determined from the identity of the SLAP reagent, so we can simplify the problem to these two components.
Having a two component reaction (with no variation in conditions such as temperature, solvent, etc.), our data set has two dimensions where a dependency exists. Consider the following four questions we may ask a machine learning model:
- “For this reaction, that was in your training data, what will be the outcome?”
- “For this reaction, where both components were in your training data, but not in this combination, what will be the outcome?”
- “For this reaction, where on of the two components was in your training data, what will be the outcome?”
- “For this reaction, where none of the components was in your training data, what will be the outcome?”
These questions get progressively more difficult. It should also be clear that question 1 (Q1) allows the model to cheat by just memorizing individual outcomes so this is not a question we should ask when evaluating models. Questions 2–4 are all legitimate questions, but they will all give different success rates. In the paper, we formalize these questions as the 0D split (Q2), 1D split (Q3), and 2D split (Q4) (ND means N independent dimensions here). When we ask, “is this model useful?”, we have to be aware which of these three questions will be asked when the model is used.
We found that we can produce useful models for the 0D split and the 1D split easily, and that for the 2D split things are more complicated. However, as long as the query molecule has some structural similarity to training compounds, the model is significantly more accurate than chance on the 2D split as well.
We ended up training different models to answer each of the questions because one of the interesting findings was that for Q2/0D split, it just does not help endowing the model with an ability to understand chemistry. Just using the dependency structure of the data set, we achieve the same outcome.
To make all this usable, we programmed an interface to the model that will take care of all this complexity for you. You just input the molecule, and the appropriate model is dispatched to predict the outcome. The prediction is accompanied by a confidence score, that reflects both the complexity of the question and the structural similarity of the query molecule to the training data.
Key points
Here we explored a few notions from the paper that deserve additional emphasis:
- Parallelizing photochemical reactions required us to build our own hardware
- Purifying and analyzing almost 500 compounds from crude reaction mixtures is possible on an automated platform
- Determining and/or predicting product properties allows for rational choice from virtual libraries
- The same can be said for synthesis prediction, but our evaluation methods need more rigor
Resources
- The paper
- All code is available on GitHub: Analysis + VL and ML code
- Repository for supplementary data, including the VL
Final words
Last but not least, it remains to say that this paper was a large collaborative effort spanning multiple years. Moritz Jackl started the project and developed the synthesis method for morpholines. Chalupat Jindakun expanded the method to piperazines and conducted many of the reactions. The Microcycle team at Novartis led by Cara Brocklehurst and Alexander Marziale was invaluable in obtaining pure compounds and a wealth of data. Clayton Springer, then also at Novartis, predicted the properties of 20 million molecules to annotate our virtual library. All of them were vital in producing this work.